Biotechnology Accelerator Division
The Biotechnology Accelerator Division identifies companies developing novel anti-cancer therapies and co-funds specific projects that will enable a company to partner or raise additional funding to complete the testing, registration and marketing of new therapies or diagnostics for blood cancer indications.
For information on how to apply to TAP, please click here.
Clinical Studies Ongoing
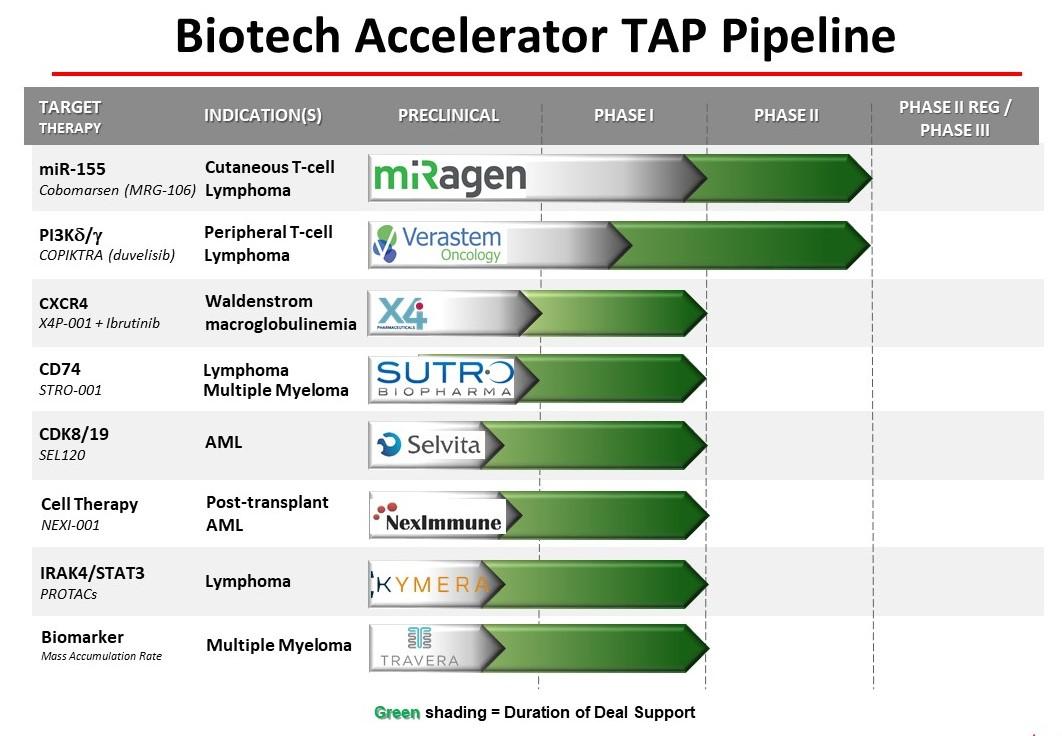
miRagen
SOLAR: A Phase 2, Randomized, Open-label, Parallel-group, Active Comparator, Multi-center Study to Investigate the Efficacy and Safety of Cobomarsen (MRG-106) in Subjects With Cutaneous T-Cell Lymphoma (CTCL), Mycosis Fungoides (MF) Subtype
The main objective of this clinical trial is to study the efficacy and safety of cobomarsen (also known as MRG-106) for the treatment of cutaneous T-cell lymphoma (CTCL), mycosis fungoides (MF) subtype. Cobomarsen is designed to inhibit the activity of a molecule called miR-155 that may be important to the growth and survival of MF cancer cells. The study will compare the effects of cobomarsen to vorinostat, a drug that has been approved for the treatment of CTCL in the United States, Canada, and Australia.
Participants will be randomly assigned to receive either weekly doses of cobomarsen or daily doses of vorinostat. Participants will continue on their assigned treatment as long as their disease does not get worse or as long as they do not have unacceptable side effects. The effects of treatment will be measured based on changes in skin lesion severity, disease-associated symptoms, and quality of life, as well as the length of time that the subject's disease remains stable or improved, without evidence of disease progression. The safety and tolerability of cobomarsen will be assessed based on the frequency and severity of observed side effects. Participants assigned to receive vorinostat who experience progression of their disease during their participation in this study may have the option to be treated with cobomarsen in a separate clinical trial, if they meet the entry criteria for that study.
ClinicalTrials.gov identifier: NCT03713320
Visit the miRagen website for additional information.
Verastem, Inc.
A Phase I Trial of Duvelisib in Combination With Either Romidepsin or Bortezomib in Relapsed/Refractory T-cell Lymphomas
The phase I portion of the study is designed to determine the MTD of duvelisib with romidepsin and duvelisib with bortezomib. All patients must have a relapsed or refractory T-cell lymphoma. The design is a standard 3+3 dose escalation of two parallel phase I studies. Patients will be enrolled in Arms A (Romidepsin + duvelisib) and B (Bortezomib + duvelisib) of the study starting at dose level 1.
Three to six patients will be initially treated in each dose level until the MTD is determined. If dose level 3 is achieved without exceeding one dose limiting toxicity (DLT) after 1 cycle that dose level will be deemed the "optimal dose" and the study will proceed to the cohort expansion phase. Should patients be found to develop significant toxicity or not tolerate therapy at the MTD with repeated cycles of therapy, the "optimal dose" may be determined to be at dose levels less than the MTD. Patients with measurable disease treated in the phase I at the optimal dose will be counted towards the accrual of the expansion cohorts by disease subtype and all efficacy and toxicity endpoints.
Cohort Expansion Phase: The expansion cohorts will further assess toxicity and safety and allow a preliminary assessment of the efficacy of the combination to provide background for a potential future subtype specific phase II study. The assessment of efficacy will be descriptive.
ClinicalTrials.gov identifier: NCT02783625
Visit the Verastem website for additional information.
X4 Pharmaceuticals, Inc.
A Phase 1b Trial of Mavorixafor, an Oral CXCR4 Antagonist, in Combination With Ibrutinib in Patients With Waldenstrom's Macroglobulinemia (WM) Whose Tumors Express Mutations in MYD88 and CXCR4
The primary objective of the study is to establish a pharmacologically active dose of mavorixafor in combination with ibrutinib based on pooled safety, clinical response, pharmacokinetic (PK) and pharmacodynamic (PD) data to select the recommended dose for a randomized registrations trial.
This is an intrapatient dose-escalation study. Three dose levels of mavorixafor will be explored: 200 milligrams (mg) once daily (QD) (dose level 1), 400 mg QD (dose level 2), and 600 mg QD (dose level 3). Ibrutinib will be administered at its labeled dose for participants with WM, 420 mg orally QD. Each treatment cycle will be 28 days.
Mavorixafor is a first-in-class, oral, small molecule allosteric antagonist of the chemokine receptor CXCR4 and is designed to address certain rare primary immunodeficiency diseases and certain cancers, including lymphomas, in which genetic mutations in CXCR4 create abnormal trafficking of white blood cells and play a role in disease process.
ClinicalTrials.gov identifier: NCT04274738
Visit the X4 Pharma website for additional information.
Sutro
Study of STRO-001, an Anti-CD74 Antibody Drug Conjugate, in Patients With Advanced B-Cell Malignancies
This study is a first-in-human Phase 1, open-label, multicenter, dose escalation study with dose expansion to identify the maximum tolerated dose (MTD), the recommended phase 2 doses (RP2D) and to evaluate the safety, tolerability, and preliminary anti-tumor activity of STRO-001 in adult subjects with B-cell malignancies (MM and NHL) who are refractory to, or intolerant of, all established therapy known to provide clinical benefit for their condition (i.e., trial subjects must not be candidates for any regimens known to provide clinical benefit). The study will consist of two parts: Part 1, dose escalation, and Part 2, dose expansion.
During Part 1 (dose escalation), an accelerated dose titration design will be applied to cohorts A (MM) and B (NHL). Doses will be escalated using an N-of-1 until the first instance of a treatment-related, clinically relevant Grade 2 non-hematologic toxicity or a Grade 3 hematologic toxicity of any type is observed during Cycle 1 (first 28 days). Any event meeting these criteria will be reviewed and confirmed by the Safety Evaluation Team (SET). Each dose escalation cohort will be assessed independently. When these criteria are met then the dose is expanded with 2 additional subjects and the standard 3+3 trial design is used for all further dosing cohorts. The dose escalation (Part 1) phase of the study will be complete when the MTD is determined and the recommended dose for Part 2 (dose expansion) is identified. The RP2D will be selected based on the safety, tolerability and exposure of STRO-001, and will be the end of Part 1 of the study. After determination of the RP2D, subjects with MM or NHL will be enrolled into indication specific dose expansion cohorts (Part 2). The accelerated dose titration (N-of-1) design with seamless transformation into a traditional 3+3 design allows for very low starting doses to be evaluated in fewer patients.
ClinicalTrials.gov identifier: NCT03424603
Visit the Sutro website for additional information.
Selvita S.A.
A Phase Ib Study of SEL120 in Patients With Acute Myeloid Leukemia or High-risk Myelodysplastic Syndrome
SEL120 is an ATP-competitive and selective inhibitor of CDK8 and a closely related serine kinase, CDK19. CDK8 is a part of a multi-protein complex that regulates gene expression and is distinct from CDK4 or CDK6, which play a role in cell cycle progression. In laboratory experiments, treatment with SEL120 has been shown to result in the death of AML cells especially with elevated phosphorylation of STAT5 and stem cell characteristics, which is significant because AML stem cells are typically resistant to conventional therapies and thereby mediate relapsed disease. Moreover, the status of phosphorylation of STAT5 may provide a useful biomarker for action of the drug. The molecular mechanism of action involves modulation of various oncogenic transcriptional programs that are critical to the survival of AML cells. This unique mechanism, which does not overlap with existing therapies, may allow the development of highly effective combination therapies that may be required to provide long term control of AML in patients.
This first-in-human study will evaluate SEL120, a novel small molecule CDK8/19 inhibitor, in patients with Acute Myeloid Leukemia (AML) or High-risk Myelodysplastic Syndrome (HR-MDS), in terms of selection of the recommended dose for further clinical development and assessment of safety, tolerability, preliminary anti-leukemic activity, as well as pharmacokinetic and pharmacodynamic profiles. The first part of the study consists of dose-escalation cohorts where patients will receive ascending doses of SEL120 to determine the recommended dose (RD) for further clinical development. The second part of the study is an enrichment cohort where additional 6 to 20 patients will be treated with SEL120 to support the evaluation of the RD.
ClinicalTrials.gov identifier: NCT04021368
Visit the Selvita website for additional information.
NexImmune Inc.
A Phase 1 / 2 Study to Evaluate the Safety, Tolerability and Initial Anti-Tumor Activity of Adoptively Transferred T Lymphocytes Targeting WT1, PRAME and Cyclin A1 AML or MDS Patients With Relapsed Disease After Matched Allogeneic HCT
This Research study is being done to characterize the safety, tolerability, and preliminary antitumor activity of the NEXI-001 T cell product (a new experimental therapy), which contains populations of CD8+ T cells targeting multiple leukemia associated antigen peptides in patients with Acute Myeloid Leukemia (AML) or Myelodysplastic Syndrome (MDS) who have relapsed disease after an allogeneic hematopoietic cell transplant (HCT).
The study will enroll AML or MDS patients who have either Minimal Residual Disease (MRD) or relapsed disease after a human leukocyte antigen (HLA)-matched allogeneic HCT. Patients who have had an HLA-mismatched or haploidentical allogeneic HCT will not be eligible to participate in this study. Eligible patients for this study must also have ≥ 50% T-cell chimerism from the original donor at the time study entry.
The enrolled patients will undergo bridging therapy for the purposes of disease control while the NEXI-001 T cell product is being manufactured. Choice of bridging therapy administered will be per the Investigator's discretion, but is limited to acceptable agents as specified in the protocol. Bridging therapy will be administered prior to lymphodepleting (LD) therapy, with the last dose of the bridging therapy administered ≥ 14 days prior to initiation of LD therapy. Within 72 hours after completing LD therapy, patients will receive a single IV infusion of the NEXI-001 T cell product.
ClinicalTrials.gov identifier: NCT04284228
Visit the NexImmune website for additional information.
Other Active Programs
Kymera Therapeutics
Kymera‘s lead program targets IRAK4, a protein known to play a significant role in inflammation mediated by toll-like and IL-1 receptors. Kymera is planning to advance its IRAK4 degrader program in a variety of autoinflammatory and autoimmune diseases, as well as in precision-medicine targeted oncology indications. The company is also developing novel protein degrader therapies to target STAT3, an un-drugged oncogenic transcription factor as well as a driver of inflammation and fibrosis, in a range of cancers and chronic diseases. Kymera announced the closing of a $102 million Series C financing in March 2020, where The Leukemia & Lymphoma Society‘s Therapy Acceleration Program® (TAP) made a strategic investment directed toward advancing the company’s work to treat blood-based cancers.
Travera LLC
Travera’s unique technology is based on a new invention and a new biological discovery about cancer cells.
The new invention is a micro-electromechanical (MEMS) device that can weigh individual cancer cells with exquisite accuracy. This device, invented at MIT and called the Suspended Microchannel Resonator (SMR), flows cancer cells through a tiny cantilever (a diving board containing a fluidics channel) and measures the change of the resonant frequency of the cantilever with a precision of one part in a billion. This precise frequency measurement enables the ability to calculate the weight of the cancer cells with sub-picogram accuracy.
Travera is studying the use of its test by collecting bone marrow (BM) aspirate samples from patients with relapsed refractory multiple myeloma (RRMM) prior to the start of a new treatment regimen for the purposes of prospectively measuring mass accumulation rate (MAR) as a biomarker of patient response to that regimen. Travera is working with leading cancer experts at prominent academic institutions to enroll study participants.
The primary study objective is to explore whether the MAR biomarker can predict patient response in RRMM patients. In order to enable this primary objective, two patient groups will be required. First, a small number of patients with recently diagnosed with multiple myeloma and never treated (Vanguard cohort) will be enrolled to help define drug concentrations used for testing. A second larger group of patients with RRMM will then be enrolled and serve as the primary study cohort. Data will be collected to estimate the biomarker’s predictive properties and to support improvement of the MAR biomarker through additional research and discovery based on the data collected during the study.
ClinicalTrials.gov Identifier: NCT03777410
Criteria/How to Apply
When a company seeks funding, it needs to demonstrate the compounds or biologics in development:
-
Are highly innovative
-
Have therapeutic potential in blood cancer field with near-term probability of providing benefits to patients
A company also needs to demonstrate:
-
Competency of its management and scientific staff
-
Financial strength
-
Freedom to operate, including intellectual property protection
-
Well-designed regulatory and commercialization strategies
TAP builds business alliances and collaborations with companies. The funding TAP provides is not a grant, nor for building the company's infrastructure. It is designed to complement the company resources to accelerate the development of a therapeutic asset.
The TAP team reviews all companies that make an inquiry but will only invite those deemed to meet diligence criteria to complete a proposal template and be considered for TAP funding.
To submit an inquiry for initial non-confidential review, please click here to download the form.
Send a completed inquiry form and non-confidential presentation to Dr. Jun Xu, Executive Director, Therapy Acceleration Program®, The Leukemia & Lymphoma Society Mail Center, 1201 15th Street N.W., Suite 410, Washington, D.C. 20005 ; [email protected].