Discovery Links Molecular Pathways for Therapeutic Possibilities In MPN
Erik Nelson and Anne-Marie Silvi
September 3, 2019

Cancer results from the malfunction or dysregulation of normal cellular processes. The dysregulation of two such processes (or pathways) that are particularly significant for cancer development are epigenetic and metabolic changes. Epigenetic changes are reversible modifications to DNA and surrounding proteins that affect the regulation of gene activity and may be related to cellular metabolism, which are chemical reactions that process cellular components for energy, signaling intermediaries, and for construction of macromolecules. A recent publication in Cancer Discovery establishes a new link between these two pathways in the development of myeloproliferative neoplasms (MPNs), leading to the discovery of several potential therapeutic options for further exploration. This work is led by Jian Xu, Assistant Professor at the Children’s Medical Center Research Institute at UT Southwestern. Drs. Zhimin Gu and Yuxuan Liu are co-first authors on this publication.
Epigenetic Factors of MPN
The enzyme EZH2 is an agent of the epigenetic process that represses genes not needed for particular cells. Depending on the context and cancer type, mutations that either activate or inactivate this enzyme can be oncogenic drivers. Mutations of the enzyme NRAS – part of a separate signaling process that serves to mediate growth and survival signals – are also present in many cancers.
Dr. Xu reasoned that mutations in NRAS and EZH2 may cooperate to form a myeloid disease, so his team combined E2-KO (removal of EZH2 by gene knockout) with G12D (an activating mutation in NRAS) in mice. While G12D alone causes a chronic pre-cancerous MPN-like disease with a long latency, and E2-KO alone shows minimal effects, the combination (G12D/E2-KO) results in an aggressive disease that significantly shortens the life span. This disease resembles an aggressive MPN called primary myelofibrosis (PMF). Further experimentation shows that only intermediate levels of the protein complex containing EZH2, when in combination with G12D, result in disease. This demonstrates that, in the context of G12D and EZH2 loss, cancer exists only when the activity of the EZH2-containing complex is at precise levels; therefore, altering these levels may be a therapeutic strategy. EZH1 is closely related to EZH2. Deletion of EZH1 alone affects neither normal hematopoiesis nor the MPN-like disease caused by G12D. When both EZH1 and EZH2 are deleted with G12D, the onset of disease is significantly delayed compared to G12D/E2-KO without EZH1 deletion. Consequently, loss of EZH1 in this specific context is a likely another therapeutic option that may someday be utilized in the clinic.
These results establish new mouse models that recapitulate chronic and aggressive MPNs. The cooperation between different signaling pathways promotes disease, and the severity of this effect depends on the dosage of various components of these pathways.
EZH2 and Amino Acid Metabolism
To understand how EZH2 loss promotes cancer formation in the context of G12D, Dr. Xu performed gene expression analysis. As expected, G12D/E2-KO results in the modulation of expression of many genes. One of the most significantly upregulated genes was BCAT1, which is essential for the metabolism of branched-chain amino acids (BCAAs). BCAT1 catalyzes the conversion of BCAAs to branched-chain alpha keto acids (BCKAs), as well as the reverse conversion – of BCKAs to BCAAs. In normal cells, the BCAT1 gene is regulated by the H3K27me3 repressive marks, which are added by EZH2. The H3K27me3 marks close the chromatin, preventing activating histone modifications from occurring, and this blocks the recruitment of protein complexes that mediate gene expression (including the modulation of BCAT1 expression). Therefore, in G12D/E2-KO cells, with the loss of EZH2 comes a loss of the H3K27me3 repressive marks, causing an increase in the transcription of the BCAT1 gene.
Potential Therapeutic Strategies Uncovered by Elucidation of BCAT1 Activity in MPNs
Prior studies examined the correlation of BCAT1 with disease formation in the context of EZH2 deficiency, but Dr. Xu’s study explores this correlation in the more specific context of G12D/E2-KO mice. His team set out to directly test the correlation between BCAT1 expression and EZH2 loss in MPN and found that when G12D/E2-KO mice are given a BCAT1 knockdown construct, the mice have a significant delay in disease onset compared to a control group. These results directly demonstrate that BCAT1 in G12D/E2-KO mice is a crucial factor of increased disease development. Dr. Xu’s team also used two small molecule inhibitors of BCAT1 enzymatic activity and show that they reduce disease burden and prolong survival in G12D/E2-KO mice. Therefore, modulation of BCAT1 has the potential to become a therapeutic option, once it has been thoroughly investigated.
As mentioned, BCAT1 catalyzes both the conversion of BCAAs to BCKAs and the conversion of BCKAs to BCAAs. In the context of G12D-E2KO cells, which of these conversions is favored? In order to answer this question, and potentially to explain why disease development was delayed in G12D-E2KO cells with the BCAT1 knockdown, Dr. Xu’s team measured the levels of leucine (a type of BCAA) and KIC (a type of BCKA) to identify which side of the reaction, if either, is favored. The team labeled and measured each of these end products in G12D/E2-KO cells with BCAT1 present and found that G12D/E2-KO cells contain significantly higher concentrations of leucine and lower KIC relative to wildtype (normal) cells, or cells with either EZH2 or G12D mutation alone. These results clearly indicate that in G12D/E2-KO cells, the reaction is favored to create more BCAAs to promote cancer development. Further experimentation modulating BCAT1 confirmed that BCAAs play a direct role in disease in the context of G12D/E2-KO and shows that in this specific context, a BCAA-restricted diet may reduce disease.
Importantly, one function of BCAA is to enhance the signaling of mTOR, which is a known oncogenic factor when overactivated. To determine whether inhibition of mTOR affects disease in the context of G12D/E2-KO, Dr. Xu’s team directly inhibited mTOR. In cell culture-based experiments measuring the ability of cells to proliferate and differentiate, mTOR inhibition has minimal effects in wildtype (normal) and G12D mutant cells but profound effects on G12D/E2-KO cells. Furthermore, the effect of mTOR inhibition delays disease onset in G12D/E2-KO mouse models. These results show that, similarly to BCAT1 inhibition, mTOR inhibition significantly affects the oncogenic function of G12D/E2-KO cells. Since mTOR inhibitors are currently FDA-approved, a path to clinical translation of these results is clear.
The Role of Glutaminase: Another Potential Therapeutic Target
In addition to BCAT1, another critical component in the chemical process of converting BCKAs to BCAAs is glutamate. Dr. Xu’s team noted that glutamate is increased in G12D mice as well as in G12D/E2-KO mice, suggesting that NRAS is the important factor for glutamate levels. They experimentally determined that increased glutamate levels are due to the activity of glutaminase, which catalyzes the conversion of glutamine to glutamate and is likely more active in G12D cells. Increased glutamate then helps fuel the production of BCAAs. Therefore, the NRAS-activating mutation enhances BCAA production by increasing levels of glutamate, likely through enhanced glutaminase activity and/or increased glutamine uptake. Importantly, this also demonstrates that G12D and BCAT1 converge on the same pathway and cooperate to increase BCAAs. This cooperation engages distinct mechanisms, but both act through the BCKA-to-BCAA conversion reaction (see the figure below). This is a mechanism that helps ensure that the disease has enough BCAAs to continue its own survival and suggests that this may be a crucial vulnerability that may be exploited therapeutically to help certain MPN patients.
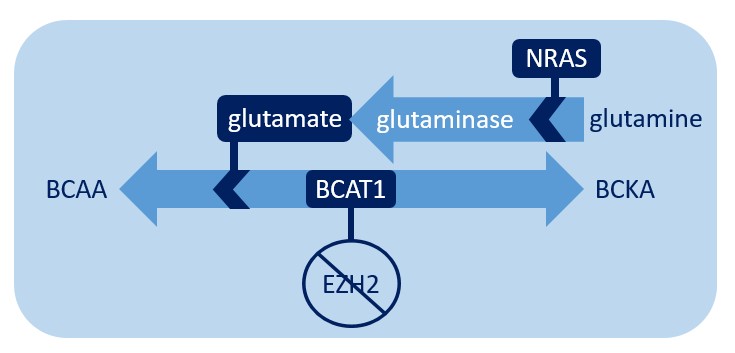
When EZH2 is deleted, BCAT1 facilitates amino acid metabolism. Glutamate, the production of which is promoted by NRAS, encourages this reaction to favor the production of BCAA.
What Does this Mean for the Future of MPN Research and Treatment?
The intersection of pathways is a recurring theme in cancer biology. Dr. Xu’s team demonstrates a direct link between epigenetics and metabolism in MPNs and points the way to various future therapeutic approaches in MPNs having inactivating EZH2 mutations that cooperate with activating NRAS mutations. Their research shows that loss of function of the EZH2 epigenetic modifier and activation of NRAS signaling converge on amino acid metabolism to increase BCAAs, leading to activation of mTOR signaling, which results in an aggressive MPN. This is important to enhance our understanding of the transformation of chronic MPNs to more aggressive disease states.
Activation of EZH2 and inhibition of NRAS proteins may prevent disease progression in this context, but there are significant difficulties in these approaches as therapeutic strategies. Other therapeutic possibilities uncovered by his work include: (1) inhibition of BCAT1, (2) use of FDA-approved mTOR inhibitors, (3) targeting of glutaminase, (4) inhibition of EHZ1, and (5) BCAA restriction in diet.
It should be noted that some of these approaches may be beneficial only in very specific disease contexts. Similar diseases caused by different oncogenic signaling pathways may not benefit from the same therapeutic approaches, and, importantly, these therapeutic approaches may be detrimental in some cases. For example, in the MPN disease described here, EZH2 loss is oncogenic; however, in other cancers an increase in EZH2 activity promotes disease. Therefore, targeted therapeutic approaches must consider the specific molecular abnormalities of that specific cancer, and patients should consult with their doctors before making any dietary adjustments. These potential therapeutic approaches need to be validated in preclinical models and rigorous clinical studies before becoming available as treatment options.
The mechanistic-based scientific discovery work conducted by Dr. Xu’s team expands our knowledge of the interplay between epigenetics and metabolism in cancer and opens up several avenues of therapeutic intervention to be preclinically evaluated.
Dr. Jian Xu is supported by The Leukemia & Lymphoma Society through a Career Development Program Scholar Award.
Reference
Gu Z, Liu Y, Cai F, et al. Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. 2019; 9(9):1228-1247.
Ananieva EA, Wilkinson AC. Branched-chain amino acid metabolism in cancer. Curr Opin Clin Nutr Metab Care. 2018; 21(1):64-70.
Click here to explore other articles on the LLS Research Spotlight.